199 SARS-CoV-2 Positively Selected Amino Acid Sites Identified That Affects Emergence Of Variants
Nikhil Prasad Fact checked by:Thailand Medical News Team Feb 27, 2024 2 years, 2 months, 1 week, 4 days, 13 hours, 9 minutes ago
COVID-19 News: The emergence of novel variants of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been a major concern since the onset of the global COVID-19 pandemic. Understanding the genetic changes that contribute to increased transmissibility or evasion of the host immune system is crucial for public health efforts. Numerous studies have employed various methods, including codeML, FEL, FUBAR, and MEME, to identify positively selected amino acid sites (PSSs) in the SARS-CoV-2 genome. However, discrepancies in results and the possibility of false positives emphasize the need for a meticulous approach.
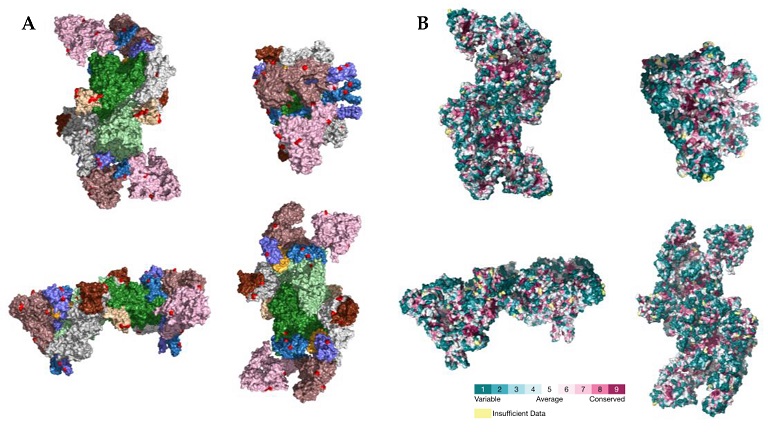 (A) Location of PSSs (labeled in red) supported by more than one type of evidence in the SARS-CoV-2 RTC (PDB accession number 7EGQ). NSP7, NSP8, NSP9, NSP10, NSP12, NSP13, and NSP14 are labeled in yellow, violet, beige, brown, green, pink, and white, respectively. (B) Consurf projection of the replicase complex dimer as in the Consurf Database. The PDB accession number 7EGQ was used as the query.
(A) Location of PSSs (labeled in red) supported by more than one type of evidence in the SARS-CoV-2 RTC (PDB accession number 7EGQ). NSP7, NSP8, NSP9, NSP10, NSP12, NSP13, and NSP14 are labeled in yellow, violet, beige, brown, green, pink, and white, respectively. (B) Consurf projection of the replicase complex dimer as in the Consurf Database. The PDB accession number 7EGQ was used as the query.
Researchers from Universidade do Porto in Portugal, Instituto de Biologia Molecular e Celular (IBMC) in Portugal, Porto University in Portugal, Faculdade de Ciências da Universidade do Porto (FCUP) in Portugal, Universidade de Vigo in Spain, and Galicia Sur Health Research Institute (IIS Galicia Sur) in Spain collaborated on a comprehensive study that is covered in this
COVID-19 News report. Their objective was to identify PSSs in SARS-CoV-2 and 15 other coronaviruses, using both codeML and FUBAR. Additionally, they aimed to compare their findings with the COV2Var database, assess the protein structures, and consider the frequency of top variants. The study provides valuable insights into the genomic evolution of coronaviruses, aiding in the identification of potential variants of concern.
Coronavirus Family and Structural Proteins
Coronaviruses, belonging to the Coronaviridae family, are known for their ability to infect both humans and animals. Among the seven human-infecting coronaviruses, including alpha and beta coronaviruses, SARS-CoV-2 has garnered significant attention due to its pandemic status. The structural proteins, namely spike (S), nucleocapsid (N), membrane (M), and envelope (E), play pivotal roles in viral structure and entry.
The M protein, crucial for nucleocapsid inclusion and S protein recruitment, exhibits an N-terminal ectodomain, a transmembrane domain, and a C-terminal endodomain. The S protein, responsible for cell tropism, consists of S1 and S2 subunits, with S1 containing the receptor-binding domain (RBD). The N protein inhibits interferon β (IFN-β) and plays a role in viral replication. The E protein, the smallest among the structural proteins, contributes to cytokine dysregulation.
Genomic
Organization and Evolutionary History
The genomic organization of coronaviruses includes structural and non-structural proteins, with the latter exhibiting lower conservation. The evolutionary history suggests the origin of alpha coronaviruses in bats and beta coronaviruses in rodents. SARS-CoV-2, initially ill-adapted to its human receptor (ACE2), underwent mutations in the S protein, enhancing its binding affinity to ACE2 and predicting increased transmissibility.
Predicting Amino Acid Variants of Interest
The ability to predict advantageous amino acid variants across the genome is vital for public health decision-making. The Centers for Disease Control and Prevention (CDC) classification system categorizes variants based on perceived threat levels. The study highlights the importance of identifying PSSs, as they indicate amino acid variants contributing to transmissibility or immune escape.
Previous studies have employed various algorithms to detect PSSs, with FUBAR revealing diversifying selection at specific amino acid sites. The COV2Var database, with data from over 13 million genomes, provides a comprehensive analysis of PSSs. However, discrepancies and potential false positives underscore the need for a rigorous approach.
Comprehensive Genome-Wide Study
The collaborative research team conducted a genome-wide study on SARS-CoV-2, utilizing both FUBAR and codeML algorithms. To enhance the reliability of their findings, they compared the results with those available in the COV2Var database. Their approach included identifying PSSs in 15 non-SARS-CoV-2 coronaviruses, considering protein structures, and evaluating the frequency of top variants.
Results and Discussion
The study identified 199 well-supported PSSs in SARS-CoV-2, shedding light on regions of the genome where amino acid variation should raise concern. The S gene exhibited the highest number of PSSs (54), consistent with its role in cell tropism and host range determination. The N protein, involved in inhibiting IFN-β, also showed a substantial number of PSSs (32).
Interestingly, non-SARS-CoV-2 coronaviruses displayed positive selection in various genes, suggesting different stages of adaptation to the host. While all genes, except nsp9, exhibited evidence of positive selection, the number of PSSs varied. This variation may be attributed to the different stages of adaptation among coronaviruses.
The researchers emphasized the importance of integrating multiple analyses, considering diverse coronavirus species, sampling schemes, positive selection detection algorithms, and protein location information. Protein regions with PSSs were identified as regions of concern, facilitating a quicker risk assessment of newly formed variants.
Conclusion
In conclusion, the comprehensive analysis of positively selected amino acid sites in SARS-CoV-2 and other coronaviruses provides valuable insights into the genomic evolution of these viruses. The identification of 199 well-supported PSSs enhances our understanding of regions where amino acid variation may impact transmissibility or immune escape. The study's approach, integrating various analyses and considering multiple sources of evidence, offers a robust framework for identifying regions of concern in other coronaviruses affecting human health. As the global community continues to monitor and respond to the dynamic landscape of viral evolution, this research contributes significantly to our ability to predict and address emerging variants effectively.
The study findings were published in the peer reviewed International Journal of Molecular Sciences.
https://www.mdpi.com/1422-0067/25/4/2428
For the latest
COVID-19 News, keep on logging to Thailand Medical News.


 Share
Share
 Tweet
Tweet
 Share
Share