BREAKING NEWS! COVID-19 Drugs: Texas Researchers Identify Extremely Potent SARS-Cov-2 Inhibitors
Source: COVID-19 Drugs Aug 01, 2020 4 years, 8 months, 3 weeks, 4 days, 6 hours, 45 minutes ago
COVID-19 Drugs: Researchers from Texas A & M University, University of Texas-Galveston and the University Of North Texas have identified a series of potent enzyme inhibitors that are able to suppress SARS-CoV-2 replication by interfering with one of the primary viral proteases.
.jpg)
The researchers urge rapid clinical and preclinical testing of their most potent compounds to meet the need for an effective antiviral therapeutics for the ongoing pandemic of COVID-19.
The study findings are published on a preprint server are pending being peer-reviewed.
https://www.biorxiv.org/content/10.1101/2020.07.28.223784v1
The SARS-CoV-2 coronavirus has a large RNA genome, almost 30 kb in size, encoding 10 open reading frames (ORFs). Among these are ORF1 and ORF1ab, which together make up over 2/3 of the entire genome and are translated into the proteins ORF1a and ORF1ab.
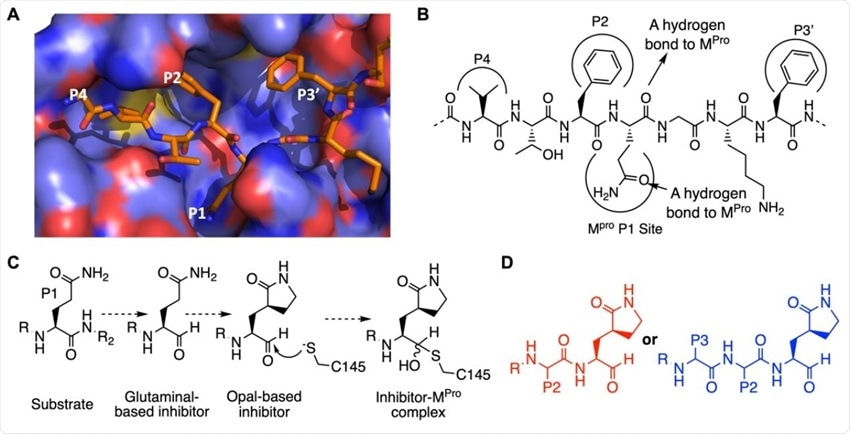 The design of SC2MPro inhibitors based on medicinal chemistry learned from SC1MPro studies. (A) The structure of SC1MPro complexed with a peptide substrate (based on the pdb entry 5b6o). Active site cavities that bind P1, P2, P4, and P3’ residues in the substrate are labeled. (B) A schematic diagram that shows interactions between SC1MPro and a substrate. (C) A scheme in which a substrate P1 residue is converted to glutaminal and then b-(S-2-oxopyrrolidin-3-yl)- alaninal (Opal) to form a reversible covalent inhibitor that reacts with the SC1MPro active site cysteine C145. (D) Scaffold structures of Opal-based inhibitors designed for SC2MPro.
The design of SC2MPro inhibitors based on medicinal chemistry learned from SC1MPro studies. (A) The structure of SC1MPro complexed with a peptide substrate (based on the pdb entry 5b6o). Active site cavities that bind P1, P2, P4, and P3’ residues in the substrate are labeled. (B) A schematic diagram that shows interactions between SC1MPro and a substrate. (C) A scheme in which a substrate P1 residue is converted to glutaminal and then b-(S-2-oxopyrrolidin-3-yl)- alaninal (Opal) to form a reversible covalent inhibitor that reacts with the SC1MPro active site cysteine C145. (D) Scaffold structures of Opal-based inhibitors designed for SC2MPro.
These two very large polypeptides undergo cleavage by the proteases to form 15 functional non-structural proteins (nsps) which are required to hijack the host cell machinery in order for the virus to express viral proteins with efficiency, replicate its genome, package the virion, and carry out transcription and genome processing efficiently to propagate the infection successfully while evading the host immune system.
This complex process is dependent on the main viral protease enzyme, SC2MPro, and therefore the inhibition of this essential post-translational cleavage enzyme could potentially be a mode of treatment of COVID-19 infection. Two regions of the polypeptides, nsp3 and nsp5, have cysteine protease activity that cleaves all the nsps.
Please Help To Donate To Sustain This Site And Other Research We Are Propelling. Thank You. https://www.thailandmedical.news/p/sponsorship
The study reports a series of compounds designed on the scaffold of SC2MPro, which have powerful inhibitory activity against this enzyme. The researchers used their knowledge of the earlier SARS-CoV-1 main protease (SC1MPro), which is 96% identical in sequence
with SC2MPro, to come up with the design of this set of compounds.
All identified compounds are tripeptidyl and dipeptidyl inhibitors and have a specialized Opal group that is capable of binding in a reversible covalent fashion with the cysteine residue 145 on the active site of SC2MPro and inhibiting it. These inhibitors are highly potent, and the half-maximal inhibitory concentration IC50 ranges from as low as 8.5 nM with the compound MP13 to 50 nM at most. This makes MPI3 “the most potent SC2MPro inhibitor that has been reported so far.”
The study team analyzed the inhibitory potency using various assays to find that these compounds were able to suppress the cytopathogenic effect (CPE) induced by SARS-CoV-2 when the latter is grown in both Vero and A549 cells. The latter is derived from the human lung epithelium and will, therefore, reflect actual human airway infection by this virus more accurately than the former.
Although MPI3 did not completely suppress CPE at all the tested concentrations, two of the compounds, namely, MPI5 and MPI8, achieved 100% suppression of the CPE at low concentrations of 2.5-5 μM in Vero E6 cells and 0.16-0.31 μM in A549 cells, respectively. The lower stability of MPI3 in the cell may explain its lower inhibition of CPE.
This could be due to the presence of Leucine and Valine at P2 and P3, which can be targeted by proteases inside and outside the cell. These cannot be changed as they optimize the enzyme structure at these sites. Still, analogs may be beneficial to maintain the high potency of inhibition while enhancing the stability of MPI3 within the cell.
The detailed crystallographic structure of the SC2MPro-inhibitor complex, formed with seven different inhibitors in this series, shows that they all form a covalent bond with C145 via the Opal aldehyde but also undergo a structural rearrangement that forms a niche for the inhibitors. MPI3 fits neatly into the binding pockets at the P1 and P2 sites of the active binding region.
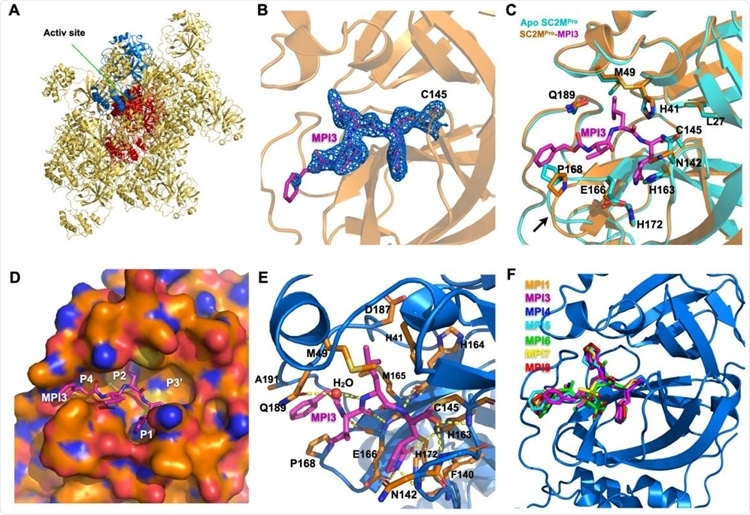 X ray crystallography analysis of SC2MPro in its apo-form and complexes with different inhibitors. (A) The packing of apo-SC2MPro in its crystals. An asymmetric unit monomer is colored in red in the center. Its active site is presented as a concaved surface. Another monomer that stacks upon the active site of the red monomer is colored in blue. (B) A contoured 2Fo-Fc map at the 1 s level around MPI3 and C145 in the active site of SC2MPro. A covalent bond between MPI3 and C145 is observable. (C) The structure overlay between apo-SC2MPro and the SC2MPro-MPI3 complex. A black arrow points to a region that undergoes structure rearrangement in the SC2MProMPI3 complex from apoenzyme to accommodate MPI3. (D) The occupation of the active site cavity of SC2MPro by MPI3. The enzyme is shown in its surface presentation mode. (E) Extensive hydrogen bonding and van del Waals interactions between SC2MPro and MPI3. The backbone of SC2MPro is colored in marine blue and side chain carbon atoms in orange. Hydrogen bonds between MPI3 and SC2MPro are depicted as yellow dashed lines. (F) The overlay of 7 Opal-based inhibitors at the active site of SC2MPro. Inhibitors are colored according to their color-coded names shown in the Figure. All images were made using the program PyMOL.
X ray crystallography analysis of SC2MPro in its apo-form and complexes with different inhibitors. (A) The packing of apo-SC2MPro in its crystals. An asymmetric unit monomer is colored in red in the center. Its active site is presented as a concaved surface. Another monomer that stacks upon the active site of the red monomer is colored in blue. (B) A contoured 2Fo-Fc map at the 1 s level around MPI3 and C145 in the active site of SC2MPro. A covalent bond between MPI3 and C145 is observable. (C) The structure overlay between apo-SC2MPro and the SC2MPro-MPI3 complex. A black arrow points to a region that undergoes structure rearrangement in the SC2MProMPI3 complex from apoenzyme to accommodate MPI3. (D) The occupation of the active site cavity of SC2MPro by MPI3. The enzyme is shown in its surface presentation mode. (E) Extensive hydrogen bonding and van del Waals interactions between SC2MPro and MPI3. The backbone of SC2MPro is colored in marine blue and side chain carbon atoms in orange. Hydrogen bonds between MPI3 and SC2MPro are depicted as yellow dashed lines. (F) The overlay of 7 Opal-based inhibitors at the active site of SC2MPro. Inhibitors are colored according to their color-coded names shown in the Figure. All images were made using the program PyMOL.
The extremely high affinity of the MPI3 compound to the enzyme is explained by strong van der Waals interactions at P1 and P2, a number of hydrogen bonds at active residues, and the covalent bond with cysteine 145. The enzyme-inhibitor complex is loosely bound, however, and further work is expected to optimize the N-terminal capping site to make it still more powerful.
The identified two most potent CPE inhibitors, MPI5 and MPI8, have Opal and Cha residues at P1 and P2 sites, the former occupying the P1 binding pocket of the enzyme and maintaining strong bonding via 3 hydrogen bonds to the lactam amide moiety of the Opal.
Significantly, the potency of viral inhibition with these compounds is markedly higher compared to several molecules that are undergoing preclinical and clinical testing. Moreover, in real life, the half-maximal effective concentration EC50 is probably much lower, since these experiments observed the lowest concentration needed to abolish CPE altogether. In fact, the SC2MPro acts reliably only at a concentration of 10 nM or more, while MPI3 has an IC50 far lower than this.
The study team says, “MPI5 and MPI8 are, as far as we know, are the most potent anti-SARS-CoV-2 small molecules in infected cells that have been reported so far.”
The study team proposes several modifications to enhance further the potency of these molecules, such as adding more heteroatoms to Opal to exploit the side-chain amide of N142 by more hydrogen bonds. Switching out the scissile amide in the enzyme inhibitor for an aldehyde will keep the hydrogen bond intact, maintaining high affinity for the enzyme, while allowing a covalent bond to be formed with Cysteine 145.
The researchers also describe the possibility of introducing a modified a-ketoamide in place of the aldehyde in the inhibitor to make use of all these potential benefits while also harnessing the binding potential of the hitherto empty P3 binding site.
A selected combination of the features of MPI3, MPI5, and MPI8 will, therefore, help generate extremely potent inhibitors.
Corresponding author, Professor Dr Wenshe Ray Liu from the department of Molecular and Cellular Medicine, College of Medicine, Texas A&M University told Thailand Medical News, “Our research indicates that there is a large chemical space that needs to be explored for the development of SC2MPro inhibitors with extreme potency. Due to the urgent matter of the COVID-19 pandemic, MPI5 and MPI8 may be quickly advanced to preclinical and clinical tests for COVID-19.”
For more on the latest research and developments on
COVID-19 Drugs, keep on logging to Thailand Medical News.
Please Help To Donate To Sustain This Site And Other Research We Are Propelling. Thank You. https://www.thailandmedical.news/p/sponsorship