BREAKING! Study Shows That A Variety Of SARS-CoV-2 Variants Are At Play In A Single Infection. Current Approach To Sequencing And Reporting Variants Is Wrong!
Source: Medical News - SARS-CoV-2 Variants Sep 09, 2022 3 years, 6 months, 3 weeks, 10 hours ago
The COVID-19 pandemic has highlighted many times over how researchers and scientists with their ‘dinosaur’ mentality and belonging to the traditional old school of thoughts have been misleading the world with their stupid ‘expertise.’
The whole approach and methods deployed to dealing with this SARS-CoV-2 crisis has been wrong from day one.
We first had COVID-19 diagnostics created by charlatans which are simply nasal or saliva swabs that are sent to RT-PCR platforms to determine whether we have SARS-CoV-2 or have recovered from COVID-19. None of these diagnostics could really assess if a person had really recovered as the virus could hide in reservoirs in the various tissues or organs of the human body and already numerous studies are showing that viral persistence is a major occurrence in most people, and is also contributing to what is misleadingly being called as Long COVID.
We had 'primates' in the beginning of the pandemic, telling us fallacies that the SARS-CoV-2 is unlikely to mutate or that even if it mutates, it will be less pathogenic and will die off. Fast forward today, we are dealing with an unprecedented variety of variants that are evolving at a very rapid rate.
Then we have the unethical and greedy lots that were only focused on antibody-based therapeutics ranging from convalescent plasma therapies to inoculations and also monoclonal or polyclonal therapeutics with claims that these could treat COVID-19 infections, prevent transmission and spread and even prevent disease severity and risk of mortality!......have yet o see any strong evidence of these claims.
We have been misled into using various drugs and therapeutics without understanding that various new variants exhibit different pathogenesis and affects the immune system in different ways and also some have extensive mutations that affects the MPro proteins, changing the way antivirals have an efficacy against them.
We have been misled by drug and therapeutic studies which do not identify the variants they are dealing with in their experiments or clinical trials!
The list can go on.
We have also been misled by the some in the scientific community that we are infected with COVID-19, a single particular variant is responsible to what was happening to our bodies.
A new study by researchers not only dispels the above but also shows that current genomic sequencing methods and ways of identifying variants have to a large extent been wrong.
The new study conducted by researchers from Case Western Reserve University, Cleveland-USA shows that all COVID-19 infections include a wide mix of SARS-CoV-2 virus variants.
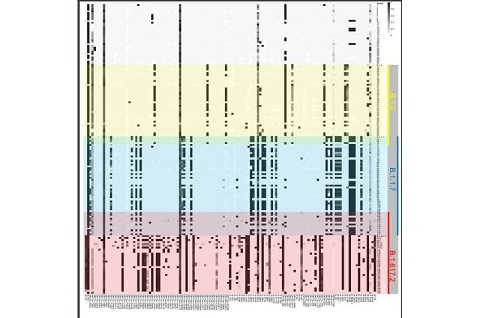
The study team presented data from over 360 patients to characterize the complex sequence diversity of individual infections identified during multiple variant surges (e.g., Alpha and Delta).
The study team observed significantly increasing SARS-CoV-2 sequence diversity during the pandemic and frequent occurrence of multiple biallelic sequence polymorphisms in all infections.
Importantly the sequence polymorphism shows that SARS-CoV-2 infections are heterogeneous mixtures.
Current convention for reporting microbial pathogen
s guides investigators to report a majority consensus sequence.
The study team found that this approach would under-report sequence variation in all samples tested.
As a result of the study team discovering that this sequence heterogeneity is efficiently transmitted from donors to recipients, their study findings illustrate that infection complexity must be monitored and reported more completely to understand SARS-CoV-2 infection and transmission dynamics.
Interestingly, many of the nucleotide changes that would not be reported in a majority consensus sequence have now been observed as lineage defining SNPs in Omicron BA.1 and/or BA.2 variants. This suggests that minority alleles in earlier SARS-CoV-2 infections may play an important role in the continuing evolution of new variants of concern.
The study findings were published in the peer reviewed journal: PLOS Genetics.
https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1010200
The most important takeaway from the study was the researchers found wide genetic variation in SARS-CoV-2 viruses among 360 patients whose viral infections were genetically sequenced, showing that all individual infections include multiple variants of the virus.
The study team noted that reporting about the virus usually highlights a single dominant strain, which leads to under-reporting virus genetic variation and can have serious consequences in public-health planning and response.
Lead author, Ernest (Ricky) Chan, director of the bioinformatics core with the Cleveland Institute for Computational Biology at the Case Western Reserve School of Medicine told Thailand
Medical News, "Our study findings bring attention to the complexity of infectious diseases that is often over-simplified when considering only the most abundant virus in an infection, and we demonstrate the importance of examining the variations that are historically considered ‘noise’.
We see that genetic variants observed in low frequency in SARS-CoV-2 infections can be early indicators of new strains responsible for later transmission surges."
The study team performed full genome sequencing of SARS-CoV-2 viruses from 250 patients in Northeast Ohio and used similar data from another 110 patients with full genetic sequences of infecting viruses provided through international research collaborators.
It should be noted that these data were developed in the early days of the COVID-19 pandemic when the Alpha variant and then the Delta variant were of major concern.
Interestingly, the study findings showed that mutations found in Omicron BA.1 and BA.2 were already present as relatively minor variations at least a year before Omicron and its many iterations became "variants of concern." Omicron and its own variants were central to a major COVID-19 resurgence last winter.
Corresponding author, Dr Peter Zimmerman, a professor in the Department of Pathology at the School of Medicine commented, "Concentration on a majority consensus of virus variants within the global research community diverts attention from genetic variation that may contribute significantly to the continuing evolution of the COVID-19 pandemic. Focus on majority variants is a critical first step in development of diagnostics, therapeutics and vaccines, however the research community needs to quantify and report out variation, so that the public health community and the general public are better prepared and nimble in response to the ever-evolving virus."
A lot of efforts are being made to define and track the emergence of virus lineages across the ongoing evolution of SARS-CoV-2 around the world. In the interest of time, global researchers have been relying on tracking and reporting on relatively dominant variations.
However, the study team noted that, given the multiple variations within single infections, it is important to report a more complete representation of the viral genetic sequences to understand how these genetic changes can spread and potentially interact with different categories of patient conditions, including evasion from eradication efforts.
Other of scientist are also worried that the current method of treating a person based on only considering the major variants and not including the minor variants could also affect disease progression and other developing medical conditions.
The study team warned,” Complex viral sequence data sets down to consensus sequences that report the majority nucleotide at each of over 29,000 positions in the SARS-CoV-2 genome. We observe that this eliminates considerable sequence variation and leads to a significant underestimation of SARS-CoV-2 infection diversity and transmission complexity. Additionally, concentration on the majority consensus sequence diverts attention from genetic variation that may contribute significantly to the continuing evolution of the COVID-19 pandemic and maybe even to preventive and treatment protocols.”
For the latest
SARS-CoV-2 Research, keep on logging to Thailand
Medical News.
Read Also:
https://www.thailandmedical.news/news/breaking-covid-19-news-ibm-study-shows-co-infections-with-various-sars-cov-2-lineages-becoming-common-heteroplasmy-more-prevalent-that-thought


 Share
Share
 Tweet
Tweet
 Share
Share