Source: Nature’s Journal Of Science, doi: 10.1038/d41586-018-05573-4 Jul 12, 2018 7 years, 9 months, 2 weeks, 1 day, 5 hours, 14 minutes ago
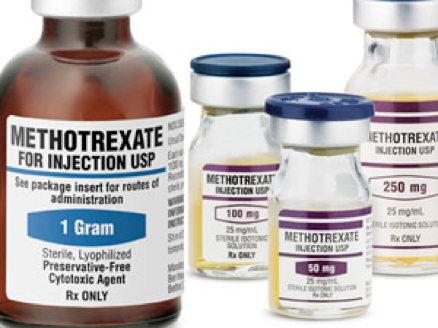
Clinical use of the anticancer drug methotrexate can be limited by its high toxicity. It emerges that a diet rich in the amino acid histidine increases the effectiveness of methotrexate treatment and lowers toxicity in mice.
Methotrexate was one of the
first approved anticancer drugs, and is a cornerstone of modern chemotherapy for the treatment of certain solid tumours and blood cancers. However, this therapy must often be stopped prematurely because of its high toxicity in patients’ healthy cells.
Writing in Nature, Kanarek
et al.1 report that degradation of the amino acid histidine can increase the sensitivity of cancer cells to methotrexate, and the results suggest that this approach can also help to lower the side effects of methotrexate treatment in mouse models.
An enzyme cofactor termed folate is required for DNA and protein synthesis. In 1948, it was found
2 that leukaemia is particularly sensitive to drugs that block folate synthesis. Methotrexate inhibits the enzyme dihydrofolate reductase (DHFR), which synthesizes the active form of folate known as tetrahydrofolate (THF) from its precursor dihydrofolate (DHF) (Fig. 1). However, when this drug is used
in the clinic, it can substantially reduce the level of folate in healthy cells — a side effect that is only partially relieved by taking folate supplements. If this toxicity reaches a level that compromises the function of healthy cells, therapy may have to be stopped prematurely. There is therefore great interest in trying to reduce the side effects of methotrexate treatment.
To investigate the processes that affect the cellular response to methotrexate, Kanarek
et al. used a cell line derived from a person with leukaemia. They employed a gene-editing technique that prevents the expression of individual genes throughout the entire genome in such cells, and tested whether a decrease in the expression of any particular gene reduced the cells’ sensitivity to methotrexate. One such gene that they identified encodes a protein called SLC19A1, which transports folate and methotrexate into cells. This result was unsurprising, given that SLC19A1 has previously been linked
3 to sensitivity to methotrexate, but it confirmed the power of the authors’ approach.
Kanarek and colleagues found that decreasing the expression of a gene that encodes the enzyme formimidoyltransferase cyclodeaminase (FTCD) also reduced the sensitivity of the cancer cells to methotrexate. FTCD degrades histidine, an amino acid that humans must obtain from their diet because the body cannot synthesize it. The authors reasoned that, because FTCD requires THF as a cofactor for its activity, a decrease in the intracellular levels of FTCD could help to limit the reduction of cellular pools of THF that occurs with methotrexate treatment. Consistent with this hypothesis, the authors fo
und that when the levels of FTCD were reduced by gene editing, THF depletion in human cancer cells treated with methotrexate was less than that seen in methotrexate-treated cells in which FTCD levels were not reduced.
On the basis of these findings, the authors predicted that the manipulation of other steps in the histidine-degradation pathway could affect the efficacy of methotrexate treatment. To test this, they again used gene editing to decrease the expression of other genes in this pathway in human cancer cells grown
in vitro. They found that such decreases did indeed mimic the effect of decreased FTCD levels in preserving the cellular pool of THF in methotrexate-treated cells.
The authors then analysed a panel of cancer cell lines, and observed a correlation between cell lines that had low expression of the enzyme histidine ammonia lyase (HAL), which acts at the first step in the histidine-degradation pathway, and low sensitivity to the effects of methotrexate. Moreover, when Kanarek and colleagues analysed data
4 available for people who have a form of leukaemia that is commonly treated with methotrexate, they found that patients who had high levels of HAL expression had higher survival rates than those with low HAL expression levels. Perhaps this is because methotrexate is more effective when high levels of HAL boost histidine degradation and the depletion of THF pools by FTCD.
The authors therefore proposed that greater cellular histidine degradation should deplete the THF pool and thus increase the effectiveness of methotrexate. To explore this prediction, they generated tumours in mice, and gave the animals methotrexate and a histidine-rich diet. They treated the animals with lower doses of methotrexate than those normally used in such models, to assess whether histidine had a synergistic effect in this context. The histidine-rich diet did indeed boost tumour-growth inhibition in response to low-dose methotrexate compared with the effects of methotrexate in animals that did not receive a dietary histidine boost.
An unexpected and exciting finding of Kanarek and colleagues’ work is that, in their mouse models, the toxicity that is usually seen in non-cancerous tissues with methotrexate treatment did not occur when the animals were given a histidine-rich diet, despite the extra toxicity that would have been expected as a result of the depletion of THF in these tissues. The results not only indicate that histidine can increase methotrexate-driven cytotoxicity in cancer cells, but also raise the possibility that some mechanism is reducing the toxic effects of THF depletion in normal tissues. Do normal and tumour tissue differ in sensitivity in this context as a result of having different thresholds for toxicity, or is the rate of histidine degradation different in normal and tumour tissues?
Given the technical challenges involved in measuring certain intermediate molecules in the histidine-degradation pathway, histidine’s contribution to the folate pool still needs to be quantified
in vitro and
in vivo to validate the authors’ model. Furthermore, there are other pathways
5, in addition to that of histidine degradation, for example degradation of the amino acid methionine, that can affect the THF level in cells. It will be important to investigate the extent to which such pathways contribute to the THF pool, and whether they also affect methotrexate-associated toxicity.
Kanarek and colleagues’ work indicates the potential value of conducting clinical trials to investigate whether histidine supplementation can boost the effectiveness of methotrexate in cancer treatment. If this is the case, further clinical trials would be needed to determine the optimal dosage. Histidine supplementation might provide an inexpensive and safe strategy for increasing the efficacy of methotrexate. Supplementation with low doses does not have adverse effects in humans, although doses of more than 8 grams of histidine a day can lead to zinc deficiency
6. However, there have been no extensive studies to investigate possible detrimental effects of a histidine-rich diet in humans.
Finally, these results might have relevance for people with rheumatoid arthritis or other chronic autoimmune diseases, in which low-dose methotrexate is the standard treatment
7 and folate supplements are the only strategy currently available to limit side effects. Could a histidine-rich diet reduce side effects in these people also, and increase their well-being?
Reference: Nature’s Journal Of Science,
doi: 10.1038/d41586-018-05573-4