Source: PAREXEL Nov 12, 2018 6 years, 5 months, 2 weeks, 12 hours, 56 minutes ago
After two chimeric antigen receptor (CAR) T-cell therapies won FDA approval in 2017 to treat children and young adults with acute lymphoblastic leukemia (ALL) and adults with diffuse large B-cell lymphoma, the American Society of Clinical Oncology hailed CAR T-Cell Immunotherapy as the Advance of the Year.1 Complete remission/response rates in clinical trials for these medicines (both targeting the CD19 antigen on B cells) were unprecedented— ranging from 54% to 81%—and clinically robust considering expected outcome from other available treatment options for these patients.2,3
More than 100 companies are now working on CAR T-cell therapies—primarily for hematologic cancers, but also for certain solid tumors—and there has been an explosion in the number of clinical trials registered each year, from 12 in 2012 to 118 in 2016 and as of September 2017, there were 420 active trials.
4
Few companies possess all the necessary in-house skills to develop CAR T-cell products, which are associated with significant challenges, due to the following factors, among others;
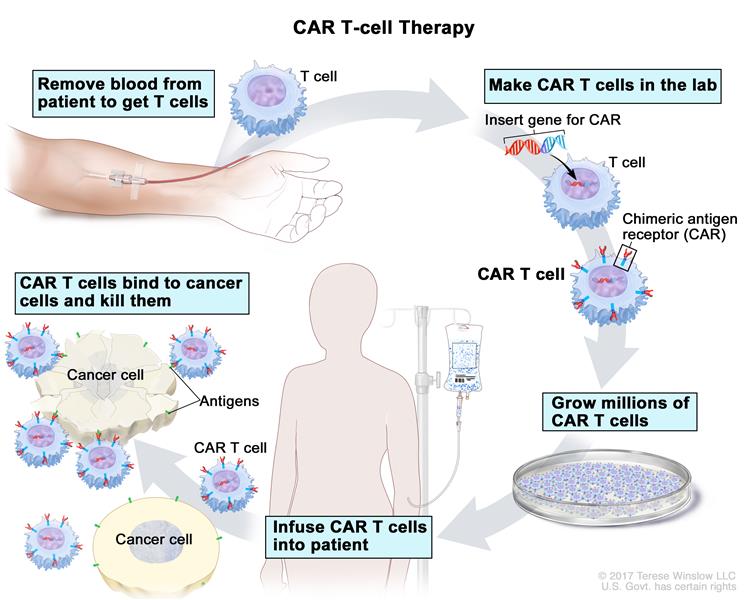
• CAR T-cell therapies require harvesting T cells from the patient that is transported to a manufacturing site, modified, and then administered back to the patient often before batch release quality control testing can be done (something unheard of for a conventional therapy);
• It’s hard to find and manage investigational sites that have the right experience and expertise to handle and administer CAR T-cell therapies;
• For some patients, CAR T-cell therapies can cause serious side effects, including a deadly cytokine storm (a severe inflammatory syndrome producing hypotension, vascular leak, pulmonary edema and coagulopathy resulting in multi-organ failure).
• Expanding use of CAR T-cell treatments beyond B-cell hematological malignancies to solid tumors may prove difficult due to the heterogeneity of solid tumors, lack of tumor penetration by engineered T cells and challenges in obtaining unique tumor antigens via plasmapheresis (despite these obstacles, many companies globally are attempting to treat solid tumors).
Whether companies build up their capabilities internally or augment with external expert resources, four areas of focus are keys to successful development of CAR T-cell products.
#1. OPTIMAL DOSING
Unlike conventional drug development, setting the dose and dosing schedule for testing of CAR T-cell therapies is an inexact science owing to numerous sources of variability that include multiplicity of infection, rate of expansion and cell viability and lack of a clear dose response relationship. The result is a rather broad dosing recommendation; for example a single-dose unit dose of Kymriah (tisagenlecleucel), the first-ever approved antiCD19 CAR T-cell therapy, contains 0.2 to 5.0 x 106 CARpositive viable T cells per kilogram (kg) of body weight for patients 50 kg or less, or 0.1 to 2.5 x 108 CAR-positive viable T cells for patients more than 50 kg. Nevertheless, it is important to find the optimal dose range in order to maximize efficacy and reduce adverse events, in particular the risk of cytokine storm.
One strategy to deliver an efficacious dose while reducing toxicity is to divide the dose and administer it on alt
ernate days instead of as a single dose. This is the approach that Novartis took in developing Kymriah. When Novartis and competitor Juno Pharmaceuticals both started their CAR T-cell programs for ALL, Juno appeared to be ahead with its product, JCAR015. However, the FDA put Juno’s Phase II clinical trial on hold in July 2016, after treatment-related cerebral edema resulted in five patient deaths. (In a preliminary investigation of the incident, Juno concluded that individual patient characteristics and “product variability” made for a lethal combination that likely led to the fatal brain swelling.
5 ) Meanwhile, Novartis modified its dose administration schedule by dividing the original dose in two, which proved effective in mitigating toxicities. Novartis won approval for Kymriah in August 2017, while Juno has estimated that its first biologics license application (BLA) filing for a different CAR T-cell product, JCAR017 to treat B-cell Non-Hodgkin lymphoma, won’t be completed until the second half of 2018 (development of JCAR015 was terminated
6 ).
#2. LOGISTICAL EXCELLENCE
Conducting clinical trials for CAR T-cell therapies is substantially more complex than for most other types of oncology therapeutics. The treatment plan involves:
• Harvesting T cells from patients in the clinic (leukapharesis);
• Washing the T cells to separate them from the leukocytes; • Sorting and separating them using specific antibody bead conjugates or markers;
• Genetically engineering the T cells using a disarmed virus so the T cells produce CARs on their surface: the special receptors that allow the T cells to recognize and attach to a specific protein;
• Expanding the modified T-cells and then cryopreserving them;
• Thawing and infusing them back into the patient.
Clinical trial personnel and sites must be able to manage the transfer of cellular products from the site to a central facility and their return to the site. During the transfers, chain-of-custody coordination and failsafe chain-ofidentity processes must be maintained, with preparation, labelling, paperwork, and shipping all performed under temperature-controlled conditions.
Far more rigorous process validation and controls are needed for CAR T-cell therapies than for conventional products because each batch of product is unique to the patient (for autologous products, ensuring the return of transfected product to the correct recipient is clearly essential). The primary goal of any effective logistics operation is to create a system that makes sure that the right product of the right quality is given to the right patient at the right time.
Clinical research associates (CRAs), investigators and pharmacy staff should either be experienced in cellular therapies or receive intensive, specialized training. Because logistics will vary from study to study, there will always need to be careful, study-specific logistics training,
even for experienced staff. The need for training support and expertise in the specific treatment plan of a CAR T-cell therapy will continue even after marketing authorization, and will be especially critical for small- and medium size developers of these products to succeed.
Finally, study sponsors need contingency plans for lost or time-expired patient samples as freezing the patients’ cells is usually performed by the manufacturer and not at the site where cells are harvested.
#3. MULTIDISCIPLINARY DEVELOPMENT
Clinical development of CAR T-cell therapies requires companies to assemble a collaborative and integrated team of experts to span the complete process from cell harvesting through processing and subsequent administration to the patient. The point of treatment must be integrated with the technology and connected to manufacturing.
Chemistry, Manufacturing and Controls (CMC) should be embedded in CAR T-cell clinical work in such a way that makes process validation and control a priority much earlier in the product lifecycle than ever before. Process validation and compliance with current Good Manufacturing Practices (cGMP) are critical. This makes it advisable to have CMC experts at the table from day one to address product manufacturing and quality issues such as ensuring process consistency and optimizing in-process controls and batch release procedures, including capturing metrics such as transduction efficiency, potency, variability of the apheresis cells, stability, and interactions with any costimulatory domains.
Regulatory expertise also is required for global development of these therapies since regulatory requirements are not only complex but differ from country to country (even different countries within the EU apply different regulations).
In the United States, CAR T-cell therapies are overseen by multiple intersecting regulatory authorities, including the FDA’s Center for Biologics Evaluation and Research (CBER), the National Institutes of Health’s Recombinant DNA Advisory Committee (RAC) and Institutional Biosafety Committees (IBCs) at the site level.
In the EU, genetically modified organism (GMO) legislation can require registration. One difficulty is that the member state
organizations that register GMOs can be different from the competent health authority for that member state. If the vector used is replication-incompetent then the risks of release of a GMO are negligible and the impact of this extra regulatory framework can be ameliorated. Marketing approvals are via the Centralised Procedure and the Committee on Human Medicinal Products (CHMP) but involve additional scrutiny from the Committee on Advanced Therapies (CAT).
Furthermore, many CAR T-cell therapies for oncology indications qualify for special regulatory mechanisms that speed development and review times. In the United States, the 21st Century Cures Act offers expedited approval for products that win designation as a Regenerative Medicine Advanced Therapy (RMAT); in Europe, there is also the potential for conditional and accelerated approval. Regulatory experts can plan development to take full advantage of benefits such as early interactions with regulators (to discuss any potential surrogate or intermediate endpoints) and priority review—but only if these experts are made part of the development team from the earliest stages.
#4. SAFETY STRATEGY
Because CAR T-cell therapies are customized by patient, every patient may not receive the same dose. Consequently, the correlation between dose and toxicity will be difficult to evaluate, and will vary from patient to patient (autologous cells are patient-specific and each patient’s genetic make-up and immune responsiveness is unique). Therefore, sponsors will need a strategy for handling both expected toxicities (i.e., cytokine release syndrome or hypersensitivity) and unexpected toxicities during clinical trials. Due to this unique challenge, companies with limited medical resources will need a partner with robust understanding of toxicity evaluation to identify the expected range of a biologically optimal dose.
Sponsors should assume that any severe toxicity findings may lead to a clinical hold and thus a regulatory strategy to successfully remove clinical holds is required—as well as an understanding of how to operate effectively under the heightened scrutiny and more complex regulatory processes that come with CAR T-cell development.
AEs caused by CAR T-cells killing healthy tissue due to target antigen expression outside of the tumor tissue (known as off-target toxicity) need to be tracked, categorized and explained. Side effects in CAR T-celltreated patients due to cross-reactivity of the engineered antigen-binding domain with a non-related surface protein includes cytokine-release syndrome resulting in noninfective fever with elevated levels of inflammatory cytokines such as interleukin-6 and interferon-γ. For AEs, questions that must be answered include: Is it dose related? Is it truly non-infective or is there a challenge within the supply chain?
AE data must be tracked meticulously and made available to principal investigators and regulators in real time. And patients receiving CAR T-cell therapy must be tracked for years in a long-term pharmacovigilance effort to catch unintended toxicities that may emerge. (Long-term risks include insertional mutagenesis and B-cell aplasia for B-cell targeted products.) CBER mandates 15 years of follow-up for subjects who receive cellular products modified with integrating vectors.
Important questions to vet your clinical development strategy include:
• Does your medical team understand CAR T-cell therapies well enough to converse and interact with investigators on a peer-to-peer level or help overcome regulatory challenges? Or do you need a partner to help?
• Does your team have sufficient expertise to know which patients should not be enrolled in a CAR T-cell trial and to recognize atypical adverse events (AEs) that may be CAR T-cell related?
• How are you going to ensure that AEs are quickly and properly handled, including the reporting of Suspected Unexpected Serious Adverse Reactions (SUSARs)?
• Can internal company resources provide guidance regarding the choice of biomarkers and the analyses of these biomarker results or is a partner needed?
CMC regulatory teams will be required to contribute data to FDA databases for product quality. This will require establishing standards and reproducible retro/Lenti virus vectors, early establishment of a potency assay, the ability to link potency to multiplicity of infection (transduction frequency), and the means to ensure consistent expression. To highlight the complexity of reporting just one of these metrics, potency assays can involve up to 20 separate bioassays. The variability for these assays is much higher than for conventional biologics and regulators therefore generally take a conservative approach to scrutinizing them. In addition, the source of the variability must be determined (i.e., is it due to the heterogeneity of the starting cells from apheresis or to the modification/ expansion processes?).
Companies must also demonstrate the ability to establish process controls and stability limits from relatively few batch numbers, as well as the ability to predict the numerical range of key quality attributes as some of these will not be available until the product has been administered back to the patient. Regulatory authorities also require multiple tests to ensure that the viral vector is replication-incompetent and to prove that transfection has been carried out to reduce or avoid insertional mutagenesis.
GETTING CAR T-CELL DEVELOPMENT RIGHT THE FIRST TIME IS KEY
If done right, the development timeline for CAR T-cell therapies is likely to be short due to the significant, durable clinical benefit produced by engineered T cells that target oncogenic drivers of the underlying cancer. For example, Novartis’ Kymriah went from investigational new drug filing (September 2014) to FDA approval (August 2017) in fewer than three years after just one clinical trial. Getting the parallel clinical, manufacturing and regulatory streams properly aligned from the start gives companies a far better chance to be “done in one” and avoid delays.
By Bob Desai, Vice President - Technical, Keith Chidwick, Vice President - Technical, Cecil Nick, Vice President - Biotechnology and Partha Roy, Vice President - Technical, PAREXEL® Consulting
For More Info: CORPORATE HEADQUARTERS 195 West Street Waltham, MA 02451 USA +1 781 487 9900 Offices across Europe, Asia and the Americas. www.Parexel.com
References:
1. Eastman, P. (2018). ASCO Names CAR T-Cell Immunotherapy 2018 Advance of the Year. Oncology Times, 40(5), p.1.
2. Maude, S., Laetsch, T., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., Bader, P., Verneris, M., Stefanski, H., Myers, G., Qayed, M., De Moerloose, B., Hiramatsu, H., Schlis, K., Davis, K., Martin, P., Nemecek, E., Yanik, G., Peters, C., Baruchel, A., Boissel, N., Mechinaud, F., Balduzzi, A., Krueger, J., June, C., Levine, B., Wood, P., Taran, T., Leung, M., Mueller, K., Zhang, Y., Sen, K., Lebwohl, D., Pulsipher, M. and Grupp, S. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine, 378(5), pp.439-448.
3. Neelapu, S., Locke, F., Bartlett, N., Lekakis, L., Miklos, D., Jacobson, C., Braunschweig, I., Oluwole, O., Siddiqi, T., Lin, Y., Timmerman, J., Stiff, P., Friedberg, J., Flinn, I., Goy, A., Hill, B., Smith, M., Deol, A., Farooq, U., McSweeney, P., Munoz, J., Avivi, I., Castro, J., Westin, J., Chavez, J., Ghobadi, A., Komanduri, K., Levy, R., Jacobsen, E., Witzig, T., Reagan, P., Bot, A., Rossi, J., Navale, L., Jiang, Y., Aycock, J., Elias, M., Chang, D., Wiezorek, J. and Go, W. (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine,
4. Future Directions for CAR T-Cell Therapy - The ASCO Post. Ascopostcom. 2018. Available at: https://www.ascopost.com/issues/december-10-2017/future-directions-for-car-t-celltherapy/. Accessed March 13, 2018.
5. Dengler R. Cancer immunotherapy company tries to explain deaths in recent trial. Science | AAAS. 2018. Available at: https://www.sciencemag.org/news/2017/11/cancerimmunotherapy-company-tries-explain-deaths-recent-trial. Accessed March 19, 2018.
6. Juno Therapeutics. Juno Therapeutics Reports Fourth Quarter And 2016 Financial Results.; 2018. Available at: https://www.businesswire.com/news/home/20170301006434/en/ Juno-Therapeutics-Reports-Fourth-Quarter-2016-Financial. Accessed March 21, 2018.