Human Genes Play Role In COVID-19 Infections, Symptoms And Disease Progression According To International Study
Source: Human Genes Aug 27, 2020 5 years, 7 months, 6 days, 1 hour, 15 minutes ago
Human Genes: A new international collaborative study shows that human host genes also play a role in the COVID-19 infections, symptoms and also progression of the disease. The study findings were published on a preprint server and have yet to have been peer-reviewed.
https://www.medrxiv.org/content/10.1101/2020.08.21.20177246v1

COVID-19 causes a broad spectrum of symptoms from none at all to terminal respiratory or multisystem failure. Not much is known about what causes the clinical features to vary so widely. The new study shows that symptom-based prediction of the diagnosis can help identify host genes that are responsible for part of this variability.
Importantly genetic factors need to be identified so that the mechanisms of disease can be improved, and vaccines can be optimized for maximum protection. One approach to this is via genome-wide association studies (GWAS), which has been used to identify loci that define the susceptibility for many common infections. However, with the current pandemic, the low number of tests and hit-or-miss testing policies have probably resulted in the detection of a very small proportion of true positives. When only the confirmed cases are included, GWAS probably lacks the power to pick up genuine associations.
A past study reports the potential for predicting COVID-19 based on self-reported symptoms. The researchers call this the Menni COVID-19 prediction model. They examined the possibility of using such a model to facilitate the identification of genetic factors in the host, which increase the risk of developing COVID-19 symptoms “COVID-19 susceptibility” and explain the significant difference in the severity of the illness.
The data cohorts used to validate the model were the Generation Scotland, Helix, Lifelines, and Netherlands Twin Register (NTR) cohorts with 168 (0, 27, 56, and 85, respectively) positively tested COVID-19 cases with 1157 negatively tested controls.
The first step was to make sure that this model could indeed pick up cases of confirmed COVID-19 in these three different groups of patients.
The study team then calibrated the prediction model to the clinical and laboratory features found in the Lifelines cohort.
The team discovered that more healthcare workers and other essential workers were tested for the virus, and mostly younger people. More COVID-19-positive people reported contact with another infected individual.
Interestingly, symptoms like fever, anosmia, ageusia, fatigue, or cough were more common in infected people even before a positive test and continuing thereafter.
Also, potential patients, by the predictive model, showed symptom patterns similar to those with a positive test outcome. Symptom prevalence is also highest at the time of the positive test but continues afterward. Fever symptoms are less common in predicted cases compared to confirmed cases.
The study team also found that predicted cases are associated with self-reported lung disease, chronic muscle disease, psychiatric disease, cancer, and neurologic disease, but not confirmed cases relative to test negatives. However, they do not rule out the possibility of bias
in this finding.
The team used the self-reported symptom profile of 56 positive and 586 negative tested individuals to improve the accuracy of the prediction of the Menni model. Still, they found no significant difference between this model and the original one, therefore choosing to continue with the latter.
GWAS of Predicted Potential COVID-19
The study team then searched for host genetic factors that play a role in susceptibility to this infection. Finally, they validated the predicted symptoms and signs through a comparison with the results of the GWAS meta-analyses of confirmed cases. They also compared their results with those of older genetic associations with other viral diseases to look for common genetic susceptibility factors.
Significantly, they team found that two single nucleotide polymorphisms (SNPs) were linked with predicted COVID-19. They also searched for potential downstream effects of these loci. This showed enrichment for protein-protein interactions with the SLC25A6 gene.
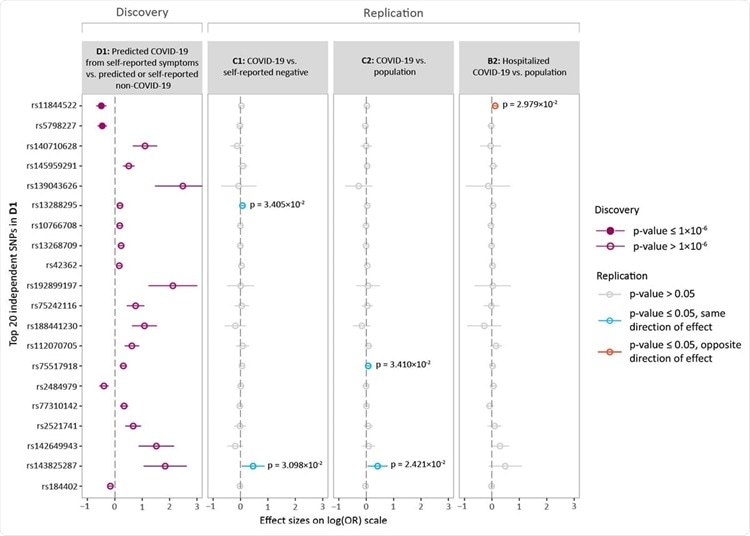 Overview of the top loci associated with predicted COVID-19. Shown are the effect size estimates of the top 20 independent SNPs associated with predicted COVID-19 (D1) and each of their associations with C1 (COVID-19 vs. self-reported negative), C2 (COVID-19 vs. population) and B2 (Hospitalized COVID-19 vs. population). The effect sizes are shown with the risk allele odds ratio (OR) on a log-scale with a corresponding 95% confidence interval (CI). Colors indicate various p-value thresholds as described in the figure legend.
Overview of the top loci associated with predicted COVID-19. Shown are the effect size estimates of the top 20 independent SNPs associated with predicted COVID-19 (D1) and each of their associations with C1 (COVID-19 vs. self-reported negative), C2 (COVID-19 vs. population) and B2 (Hospitalized COVID-19 vs. population). The effect sizes are shown with the risk allele odds ratio (OR) on a log-scale with a corresponding 95% confidence interval (CI). Colors indicate various p-value thresholds as described in the figure legend.
It should be noted that this gene encodes for a vital component of a mitochondrial system, which also takes part in apoptosis. It is suppressed in human CMV infection but is found to be expressed in apoptotic cells infected with the flu virus. This may indicate it also participates in COVID-19 susceptibility.
The study team confirmed that “self-reported disease-related symptoms are useful for prediction of infection status.” This is also the first study to report GWAS analysis on predicted potential COVID-19 in the C19HG consortium on genetic analysis of this infection.
The research findings suggests that two genetic variants are linked to predicted COVID-19, but they did not observe overlap between hits for other infections and COVID-19 phenotypes
Although the Menni model can make good predictions, it is not very sensitive, nor does it have a high positive predictive value. In other words, it misses many cases but falsely identifies others. This is an area for future research, such as the repetition of self-assessed symptom reports for better accuracy of prediction.
Also symptom sets should be precise in order to produce a narrower and specific genetic signal. If not, other conditions such as viral infections that are not COVID-19 may be predicted as potential COVID-19, rendering the GWAS less specific.
From the findings, of the top associated SNPs, one is at a genetic locus encoding immunoglobulin, and is near a gene family found to be enriched in COVID-19 patients. The replication of this SNP showed the opposite direction of effect in the B2 analysis (hospitalized COVID-19 vs population) which focuses on the severity of the condition rather than susceptibility to it.
It was also found that another SNP in the B2 GWAS analysis shows even greater significance than the top variant in the C2 analysis (COVID-19 vs. population).
Interestingly, other similar findings indicate, they say, “that the reported variants on the 3p21.31 locus are more likely to be associated with COVID-19 severity than COVID-19 susceptibility.” This means that the predicted COVID-19 phenotype cannot be used to assess susceptibility if only the lack of association with this locus is considered.
The study team says the prediction model needs to be refined since only severe phenotypes were included in the early months of the pandemic, and certain occupations were tested more frequently. Some symptoms here overlap with those of some chronic conditions which are prevalent in the population, and this may lead to a false-positive diagnosis. Different prevalence rates in different populations may also be a limiting factor.
But the study team says their findings prove the potential for GWAS studies on predicted COVID-19 cases, the latter being useful in increasing the sample case size and thus helping to uncover the genetic basis of susceptibility to the virus. This is only a proof of concept since they could not find any loci of genome-wide significance. They did demonstrate that genetic loci associated with other viral infections are not overlapping with COVID-19 susceptibility.
The team commented, “Our findings furthermore demonstrate the added value of using self-reported symptom assessments to quickly monitor the activity of novel endemic viral outbreaks in a scenario of limited testing. In case there should be another similar outbreak, they say, the collection of information on such symptoms should be carried out repeatedly over time for this purpose.”
For more articles and research about
Human Genes, keep on logging to Thailand Medical News