Japanese Scientists Uncover Pathways Involved In Lung fibrosis By Using New Mouse Model
Nikhil Prasad Fact checked by:Thailand Medical News Team Mar 10, 2024 1 year, 1 month, 2 weeks, 2 days, 8 hours, 29 minutes ago
Medical News: In the realm of respiratory medicine, idiopathic pulmonary fibrosis (IPF) stands as a daunting challenge. This progressive lung disease, characterized by the relentless scarring of lung tissue, robs individuals of their ability to breathe freely, ultimately leading to respiratory failure and death. Despite decades of research, the precise mechanisms underlying IPF remain elusive, and effective treatments are sorely lacking. However, a recent breakthrough study covered in this
Medical News report, led by scientists at the RIKEN Center for Integrative Medical Sciences-Japan offers new hope in the quest to conquer this debilitating condition.
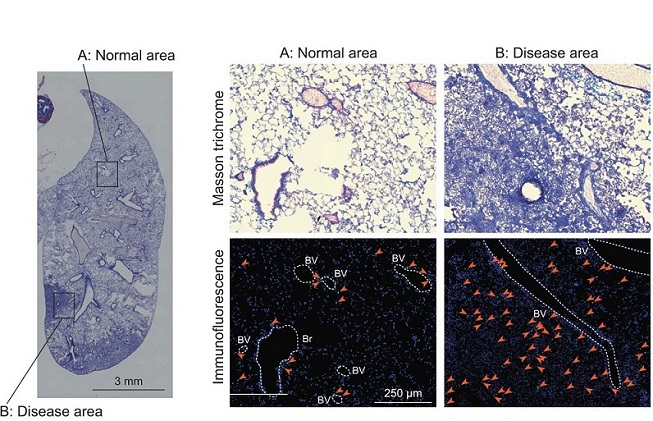 Pathways Involved In Lung fibrosis By Using New Mouse Model. MT and immunofluorescence staining images of the whole right lung lobes of Ifngr1-/-Rag2-/- mice (24 weeks; male). The areas enclosed by the squares in the left panel are enlarged and shown in the right panels (A: normal area, and B: disease area). The arrows indicate Gata3-positive cells. The dotted lines indicate blood vessels or bronchi. BV, blood vessel; Br, bronchus. Blue, DAPI; Green, Gata3. Scale bars: 3 mm (left panel) or 250 μm (right panels). Data are representative of at least three independent experiments and are presented as the mean ± s.e.m. For statistical analysis.
A Decade-Long Journey
Pathways Involved In Lung fibrosis By Using New Mouse Model. MT and immunofluorescence staining images of the whole right lung lobes of Ifngr1-/-Rag2-/- mice (24 weeks; male). The areas enclosed by the squares in the left panel are enlarged and shown in the right panels (A: normal area, and B: disease area). The arrows indicate Gata3-positive cells. The dotted lines indicate blood vessels or bronchi. BV, blood vessel; Br, bronchus. Blue, DAPI; Green, Gata3. Scale bars: 3 mm (left panel) or 250 μm (right panels). Data are representative of at least three independent experiments and are presented as the mean ± s.e.m. For statistical analysis.
A Decade-Long Journey
Nearly a decade ago, a team led by Dr Kazuyo Moro embarked on a journey to unravel the mysteries of lung inflammation and immune response. Their focus? Group 2 innate lymphoid cells (ILC2s), a specialized population of immune cells implicated in allergic reactions and lung infections. In a series of pioneering experiments, they created a mouse model lacking key immune-related genes, leading to dysregulated signaling of interferon-gamma, a vital immune molecule. While their initial aim was to understand the role of ILC2s in allergic responses, they stumbled upon a remarkable discovery - these genetically modified mice exhibited not only heightened ILC2 activity and inflammation but also the development of fibrotic lesions in their lungs as they aged.
A Model Closer to Reality
Fast forward to the present day, and the implications of this serendipitous finding are nothing short of revolutionary. Dr Moro's team has meticulously demonstrated that these mice closely mirror the progression of IPF, offering a more faithful representation than existing mouse models. Unlike traditional models where lung damage primarily originates within the airways, these mice display scarring on the lung's lining (pleural side), a hallmark of IPF pathology observed in human patients. This unique characteristic opens a window into the external triggers and progression of IPF, shedding light on the elusive origins of fibrosis in affected individuals.
Understanding the Biological Pathways
Centr
al to the progression of fibrosis in these mice is the dysregulated activation of ILC2s. Without proper interferon-gamma signaling, these immune cells become hyperactive, expressing elevated levels of a surface receptor that fosters interactions with fibroblast cells lining the outer layers of the lungs. This interaction sets off a cascade of events, leading to excessive collagen production, lung stiffness, and tissue thickening - all hallmarks of IPF progression.
Human Validation
To validate their findings, the researchers turned to human samples, isolating ILC2s from the blood of IPF patients. Remarkably, these patient-derived cells exhibited similar patterns of receptor expression and decreased interferon-gamma signaling, aligning closely with the observations in the mouse model. Furthermore, the team uncovered a positive feedback loop involving ILC2-activated fibroblasts and the production of IL-33, further fueling the fibrotic process.
Advantages and Drawbacks of the Model
While the Ifngr1-/-Rag2-/- mouse model developed by RIKEN offers unparalleled biological accuracy, it comes with its own set of challenges. Unlike other models that induce fibrosis more rapidly, these mice take approximately 15 weeks to exhibit signs of lung fibrosis. However, Dr. Moro argues that the benefits of biological fidelity far outweigh the convenience of speed, as the insights gained from this model hold immense potential for drug development and therapeutic interventions targeting IPF.
Exploring the Mechanisms Governing Fibrosis
Delving deeper into the mechanisms governing fibrosis development, the researchers have divided the disease progression into three distinct phases: intact, inflammatory, and fibrotic. This meticulous dissection allows for a nuanced analysis of pathology at each stage, offering valuable insights into the temporal dynamics of fibrosis progression - a feat not achievable with previous models.
Insights into Human IPF
One of the most striking parallels between the mouse model and human IPF lies in the progression pattern of fibrosis. While traditional models often fail to capture the pleural origins of fibrosis observed in human patients, the Ifngr1-/-Rag2-/- mice closely mimic this aspect of IPF pathology, providing a more accurate representation of disease progression. Furthermore, the activation of ILC2s during the inflammatory phase and their interaction with fibroblasts shed light on the early events driving fibrosis development, offering novel avenues for therapeutic intervention.
Role of ILC2s in Fibrosis
The pivotal role of ILC2s in fibrosis development cannot be overstated. The dysregulated activation of these immune cells, fueled by the absence of interferon-gamma signaling, sets the stage for the progressive scarring of lung tissue observed in IPF. Moreover, the direct induction of collagen expression in fibroblasts by activated ILC2s highlights the intricate interplay between immune cells and stromal elements in driving fibrosis progression.
Potential Therapeutic Interventions
Armed with these insights, researchers are now poised to explore novel therapeutic interventions aimed at targeting ILC2s during the initial stages of IPF. By modulating the dysregulated immune response underlying fibrosis development, these interventions hold promise in mitigating disease progression and improving outcomes for affected individuals.
Conclusion
In conclusion, the Ifngr1-/-Rag2-/- mouse model developed by scientists at RIKEN offers a transformative tool for unraveling the complexities of idiopathic pulmonary fibrosis. By faithfully recapitulating the progression observed in human patients, this model provides unprecedented insights into the biological pathways driving fibrosis development. From the dysregulated activation of ILC2s to the intricate interplay between immune cells and fibroblasts, each discovery paves the way for innovative therapeutic strategies aimed at halting the relentless march of this devastating lung disease. As researchers and pharmaceutical companies harness the potential of this innovative model, the future holds promise for a world where idiopathic pulmonary fibrosis is no longer an insurmountable challenge, but a conquerable foe.
The study findings were published in the peer reviewed journal: Nature Communications.
https://www.nature.com/articles/s41467-023-43336-6
For the latest on
Lung Fibrosis, keep on logging to Thailand
Medical News.


 Share
Share
 Tweet
Tweet
 Share
Share