Proteins Of Muscle Cells Targeted To Develop New Meds For Blood Pressure.
Source: University of Tennessee Health Science Center, US. Dec 06, 2018 7 years, 3 months, 2 weeks, 2 days, 22 hours, 55 minutes ago
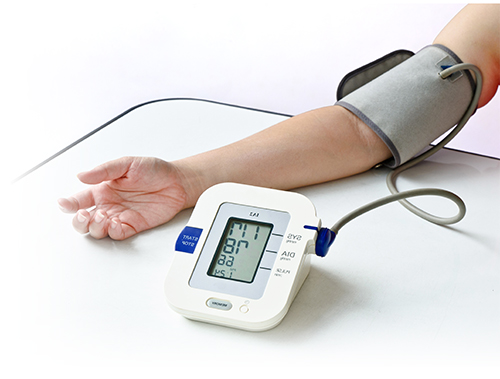
Scientists have identified a key player in blood pressure regulation and have shown that switching it off reduces blood pressure in mice.Their study ends much uncertainty about the contribution this molecule makes to high blood pressure and could lead to the development of new drugs. High blood pressure affects millions worldwide and is a leading cause of heart attack and stroke.
Blood pressure is controlled, in part, by muscle cells that line the wall of arteries. These muscle cells have proteins called Transient Receptor Potential (TRP) channels on their surface that allow sodium and calcium to move through. Around 13 different TRP channels are present on arterial muscle cells, but it is unclear if they control normal blood pressure or contribute to high blood pressure.
"The contribution of TRP channels to normal blood pressure and to changes in blood pressure are unclear, and we don't know whether the different channel types in different organs are controlled in a similar way," explains senior author Jonathan Jaggar, Bronstein Endowed Professor of Physiology at the University of Tennessee Health Science Center, US. "We chose to study a TRP channel called PKD2 because patients with genetic mutations in this protein have high blood pressure and previous research has shown conflicting results regarding its functions in arterial muscle cells."
The team switched off PKD2 in the smooth muscle cells of mice and found they had lower blood pressure than normal mice. They then looked at constriction of blood vessels (vasoconstriction) in the mice, by increasing the pressure within blood vessels of the hindlimb. They found that an increase in blood pressure increased vasoconstriction in mice with functional PKD2, but not in mice without PKD2. PKD2 did not contribute to the effects of two vasoconstrictor molecules—phenylephrine and angiotensin II.
However, by contrast, when they looked at arteries in the abdomen, they found that the PKD2 contributes to vasoconstriction stimulated by phenylephrine. This revealed that specific vasoconstrictor stimuli activate PKD2 channels in the arteries of different body tissues. In both cases, they found that PKD2 activation allows an influx of sodium into the cell which alters the electrical charge across the membrane of muscle cells and causes the vessel to constrict.
They next tested the hypothesis that removing PKD2 could reduce hypertension in mice. They gave normal and PKD2-deficient mice an infusion of angiotensin II, which elevates blood pressure, and found that the increase in arterial blood pressure was 26% smaller in the mice without PKD2. Mice with high blood pressure also had greater amounts of PKD2 on the surface of their arterial muscle cells.
Finally, the team measured the contractility of blood vessels at high pressure after treatment with phenylephrine. They found that vasoconstriction in the PKD2-deficient mice was only about 70% of that observed in mice with functional PKD2, confirming that reduced blood pressure in the PKD2-deficient mice results from blood-vessel relaxation.
"Our results indicate that stimuli which activate PKD2 channels are blood vessel specific, showing that there is not a singular mechanism regulating muscle cell contractility in all arteries," concludes Jaggar. "Our demonstration that muscle cell PKD2 channels regulate blood
pressure blood is a step forward to better understanding the importance of this ion channel target in normal cardiovascular physiology, and as a potential
drug target for cardiovascular disease."
Reference: : Simon Bulley et al, Arterial smooth muscle cell PKD2 (TRPP1) channels regulate systemic blood pressure, eLife (2018). DOI: 10.7554/eLife.42628